Понятие биоинформатики
Под биоинформатикой обычно понимают использование компьютеров для решения биологических задач. В настоящее время это почти исключительно задачи молекулярной биологии. Причина этого в том, что за последние 20-25 лет накоплен поистине колоссальный экспериментальный материал именно о строении и функционировании биологических молекул (белков и нуклеиновых кислот), в качестве примера достаточно привести геном человека. Этот материал требует развитых компьютерных методов для своего анализа. Поэтому биоинформатика в большинстве мировых научных центров понимается как синоним вычислительной молекулярной биологии.
Есть несколько основных направлений этого раздела науки, в зависимости от
исследуемых объектов:
- * Биоинформатика последовательностей.
- * Структурная биоинформатика.
- * Компьютерная геномика
Основные направления биоинформатики в зависимости от исследуемых объектов
Биоинформатика последовательностей
Биоинформатика последовательностей
Наиболее известной и наиболее эффективной областью применения биоинформатики в настоящее время является анализ геномов, тесно связанный с анализом последовательностей.
Этот раздел биоинформатики занимается анализом нуклеотидных и белковых последовательностей. В настоящее время разработаны эффективные экспериментальные методы определения нуклеотидных последовательностей. Определение нуклеотидных последовательностей стало рутинной хорошо автоматизированной процедурой. В результате рутинной хорошо автоматизированной процедуры уже получено огромное количество генетических текстов. Так, в базе данных EMBL на 15.02.2007 г. хранится 87000 493 документов с описанием нуклеотидных последовательностей, содержащих в целом 157545686001 символов (нуклеотидов), что соответствует примерно библиотеке в 105 толстых томов с убористым шрифтом. Найти нужный ген в EMBL, это все равно, что найти цитату в такой библиотеке. Без помощи компьютера сделать это, мягко говоря, очень трудно. А число данных экспоненциально растет.
Представим себе геном небольшой бактерии - это непрерывная строка длиной в 1-10миллионов символов символов, и далеко не вся ДНК кодирует белки. Первый тип биоинформатической задачи - это задачи поиска в нуклеотидных последовательностях особых участков, участков, кодирующих белки, участков, кодирующих РНК (например, тРНК), участков связывания с регуляторными белками и др. И это не всегда простые задачи, например, гены эукариотических организмов состоят из чередующихся "осмысленных" и "бессмысленных" фрагментов (экзонов и интронов), и расстояние между "осмысленными" фрагментами может достигать тысяч нуклеотидов.
Пусть ген найден. Что он кодирует? Зачем он нужен?
Если речь идет об участке ДНК, кодирующем белок, то с помощью весьма простой операции - трансляции с использованием известного генетического кода можно получить. аминокислотные (белковые) последовательности. Из известных на сегодня 4 273 512 белков около 94% последовательностей - это именно такие гипотетические трансляты, и больше о них ничего не известно. Скорость поступления информации с автоматических секвенаторов превышает скорость нашего понимания ее смысла! Но биологические объекты - это объекты, возникшие в процессе эволюции. Сравнительно-эволюционный подход - один из мощнейших подходов в биологии. Например, функция белка из одного организма хорошо экспериментально изучена, в другом организме нашли белок с похожей аминокислотной последовательностью. Можно предположить, что второй (неизвестный) белок выполняет ту же или схожую функцию. И здесь сразу возникает несколько вопросов. Во-первых, что значит похожая последовательность? Как сравнивать последовательности? При какой степени сходства последовательностей можно предполагать, что белки выполняют сходные функции? Сравнение последовательностей (выравнивание) является важнейшей задачей биоинформатики. Трудно найти современного биолога, ни разу не использовавшего программы Blastp и ClustalX, появление этих программ - уже крупный успех биоинформатики. Но современные биоинформатики недовольны и постоянно совершенствуют методы выравниваний. Можно привести много примеров того, как сравнительно-эволюционный подход в сочетании с биоинформатическими методами порождает новое биологическое знание.
Генетические тексты - тексты с большой долей шума, сравнивая родственные последовательности, в ряде случаев удается отфильтровать шум и выявить сигнал, например, короткую последовательность нуклеотидов, способную связываться с белком- регулятором, или аминокислотные остатки в ферменте, отвечающие за связывание субстрата. Чтобы быть уверенными в результате, биоинформатики используют теорию вероятности и математическую статистику. Подводя итог, можно сказать, что основные задачи биоинформатики, связанные с анализом отдельных последовательностей, состоят в следующем:
- * Выравнивание и определение сходства двух последовательностей
- * Построение множественных выравниваний
- * Распознавание генов
- * Предсказание сайтов связывания регуляторных белков
- * Предсказание вторичной структуры РНК
Создание новых экспериментальных технологий ставит перед биоинформатикой целый ряд новых задач. Например, развитие масс-спектрометрии позволяет (пока в принципе) в одном эксперименте проанализировать весь набор белков, присутствующий в клетке. Для решения этой задачи необходим совместный анализ спектров масс и геномов. Открытие новых биологических явлений и механизмов также приводит к появлению новых задач. Хорошим примером служит открытие РНК интерференции, за которую в 2006 году дали Нобелевскую премию по физиологии. Это открытие породило целый вал биоинформатических работ, посвященных поиску участков связывания микроРНК и новых микроРНК. Многие находки были затем подтверждены экспериментально.
Биоинформа́тика - в настоящее время, данным термином обозначаются любые попытки биологов ввести обобщения эвристического толка на гигантские массивы биологических данных. До недавнего времени (2000-2002г), под биоинформатикой подразумевалось, в большинстве случаев, использование процедур сравнения символьных последовательностей (аминокислотные последовательности белков, нуклеотидные последовательности ДНК и РНК). После публикации последовательности генома человека в начале 2000-х годов стало ясно, что сравнение символьных последовательностей само по себе не позволяет дать ответ на вопросы о функции генов и белков. Поэтому, сейчас происходит разворот в сторону более широкого понимания биоинформатики как "менеджмента разнородных биологических данных" (см. https://www.novapublishers.com/catalog/product_info.php?products_id=4277).
Терминология
Лексический анализ слова "биоинформатика" указывает на приставку "био-" (от греч.. Однако, как это не парадоксально, "биоинформатика" (англ. bioinformatics), до недавнего времени, не имела практически ничего общего с "информатикой" (англ. "computer science"). В этом легко убедится, проведя поиски по этим ключевым словам в базах данных публикаций по всем областям биологии, включая биоинформатику. В базе данных MEDLINE содержится почти 20 млн абстрактов (см. http://www.ncbi.nlm.nih.gov/sites/entrez). В среде биологов, под биоинформатикой понимают использование компьютеров для обработки экспериментальных данных по структуре биологических макромолекул белков и нуклеиновых кислот с целью получения биологически значимой информации. Основные усилия исследователей в этой области направлены на изучение геномов , анализ и распознавание (менее приемлемый термин "предсказание") структуры белков , анализ и распознавание ("предсказание") взаимодействий молекул белков различных типов друг с другом и др.
Термины биоинформатика и «вычислительная биология » часто употребляются как синонимы, хотя каждый автор в данной области придумывает, как правило, свои собственные определения для каждого. Иногда считают, что не всякое использование вычислительных методов в биологии является биоинформатикой, например, математическое моделирование биологических процессов - это не биоинформатика.
Основные области исследований
Анализ генетических последовательностей
Начиная с середины 1970-х, было поределено более 100 млн нуклеотидных последовательностей генов различных организмов. Эти данные используются для определения последовательностей белков и регуляторных участков. Сравнение генов в рамках одного или разных видов может продемонстрировать сходство функций белков или отношения между видами (таким образом могут быть составлены филогенетические деревья). С возрастанием количества данных уже давно стало невозможным вручную анализировать последовательности. В наши дни для поиска по геномам тысяч организмов, состоящих из миллиардов пар нуклеотидов используются компьютерные программы. Программы могут однозначно сопоставить («выровнять») похожие последовательности ДНК в геномах разных видов; часто такие последовательности несут сходные функции, а различия возникают в результате мелких мутаций, таких как замены отдельных нуклеотидов, вставки нуклеотидов, и их «выпадения» (делеции). Один из вариантов такого выравнивания применяется при самом процессе секвенирования. Так называемая техника «дробного секвенирования» (которая была, например, использована для секвенирования первого бактериального генома, Haemophilus influenzae ) вместо полной последовательности нуклеотидов даёт последовательности коротких фрагментов ДНК (каждый длиной около 600-800 нуклеотидов). Концы фрагментов накладываются друг на друга и, совмещённые должным образом, дают полный геном. Такой метод быстро даёт результаты секвенирования, но сборка фрагментов может быть довольно сложной задачей для больших геномов. В проекте по расшифроке генома человека сборка заняла несколько месяцев компьютерного времени. Сейчас этот метод применяется для практически всех геномов, и алгоритмы сборки геномов являются одной из острейших проблем биоинформатики на сегодняшний момент.
Другим примером применения компьютерного анализа последовательностей является автоматический поиск генов и регуляторных последовательностей в геноме. Не все нуклеотиды в геноме используются для задания последовательностей белков. Например, в геномах высших организмов, большие сегменты ДНК явно не кодируют белки и их функциональная роль неизвестна. Разработка алгоритмов выявления кодирующих белки участков генома является важной задачей современной биоинформатики.
Биоинформатика помогает связать геномные и протеомные проекты, к примеру, помогая в использовании последовательности ДНК для идентификации белков.
Аннотация геномов
В контексте геномики аннотация - процесс маркировки генов и других объектов в последовательности ДНК.
Основные программы сравнения аминокислотных и нуклеотидных последовательностей
- ACT (Artemis Comparison Tool) - геномный анализ
- Arlequin - анализ популяционно-генетических данных
- BioEdit
- BioNumerics - коммерческий универсальный пакет программ
- BLAST - поиск родственных последовательностей в базе данных нуклеотидных и аминокислотных последовательностей
- ClustalW
- ClustalX - множественное выравнивание нуклеотидных и аминокислотных последовательностей
- FASTA - набор алгоритмов определения схожести нуклеотидных и аминокислотных последовательностей
- JalView - редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- Mesquite - программа для сравнительной биологии на языке Java
- Muscle - множественное сравнение нуклеотидных и аминокислотных последовательностей. Более быстрая и точная по сравнению с
Может продемонстрировать сходство функций белков или отношения между видами (таким образом могут быть составлены Филогенетические деревья). С возрастанием количества данных уже давно стало невозможным вручную анализировать последовательности. В наши дни для поиска по геномам тысяч организмов, состоящих из миллиардов пар нуклеотидов используются компьютерные программы. Программы могут однозначно сопоставить (выровнять) похожие последовательности ДНК в геномах разных видов; часто такие последовательности несут сходные функции, а различия возникают в результате мелких мутаций, таких как замены отдельных нуклеотидов, вставки нуклеотидов, и их «выпадения» (делеции). Один из вариантов такого выравнивания применяется при самом процессе секвенирования. Так называемая техника «дробного секвенирования » (которая была, например, использована Институтом Генетических Исследований для секвенирования первого бактериального генома, Haemophilus influenzae ) вместо полной последовательности нуклеотидов даёт последовательности коротких фрагментов ДНК (каждый длиной около 600-800 нуклеотидов). Концы фрагментов накладываются друг на друга и, совмещённые должным образом, дают полный геном. Такой метод быстро даёт результаты секвенирования, но сборка фрагментов может быть довольно сложной задачей для больших геномов. В проекте по расшифроке генома человека сборка заняла несколько месяцев компьютерного времени. Сейчас этот метод применяется для практически всех геномов, и алгоритмы сборки геномов являются одной из острейших проблем биоинформатики на сегодняшний момент.
Другим примером применения компьютерного анализа последовательностей является автоматический поиск генов и регуляторных последовательностей в геноме. Не все нуклеотиды в геноме используются для задания последовательностей белков. Например, в геномах высших организмов, большие сегменты ДНК явно не кодируют белки и их функциональная роль неизвестна. Разработка алгоритмов выявления кодирующих белки участков генома является важной задачей современной биоинформатики.
Биоинформатика помогает связать геномные и протеомные проекты, к примеру, помогая в использовании последовательности ДНК для идентификации белков.
Аннотация геномов
Оценка биологического разнообразия
Основные биоинформационные программы
- ACT (Artemis Comparison Tool) - геномный анализ
- Arlequin - анализ популяционно-генетических данных
- BioEdit
- BioNumerics - коммерческий универсальный пакет программ
- BLAST - поиск родственных последовательностей в базе данных нуклеотидных и аминокислотных последовательностей
- Clustal - множественное выравнивание нуклеотидных и аминокислотных последовательностей
- DnaSP - анализ полиморфизма последовательностей ДНК
- FigTree - редактор филогенетических деревьев
- Genepop
- Genetix - популяционно-генетический анализ (программа доступна только на французском языке)
- JalView - редактор множественного выравнивания нуклеотидных и аминокислотных последовательностей
- MacClade - коммерческая программа для интерктивного эволюционного анализа данных
- MEGA - молекулярно-эволюционный генетический анализ
- Mesquite - программа для сравнительной биологии на языке Java
- Muscle - множественное сравнение нуклеотидных и аминокислотных последовательностей. Более быстрая и точная по сравнению с ClustalW
- PAUP - филогенетический анализ с использованием метода парсимонии (и других методов)
- PHYLIP - пакет филогенетических программ
- Phylo_win - филогенетический анализ. Программа имеет графический интерфейс.
- PopGene - анализ генетического разнообразия популяций
- Populations - популяционно-генетический анализ
- PSI Protein Classifier - обобщение результатов, полученных с помощью программы PSI-BLAST
- Seaview - филогенетический анализ (с графическим интерфейсом)
- Sequin - депонирование последовательностей в GenBank , EMBL , DDBJ
- SPAdes - сборщик бактериальных геномов
- T-Coffee - множественное прогрессивное выравнивание нуклеотидных и аминокислотных последовательностей. Более чувствительное, чем в ClustalW /ClustalX .
- UGENE - свободный русскоязычный инструмент, множественное выравнивание нуклеотидных и аминокислотных последовательностей, филогенетический анализ, аннотирование, работа с базами данных.
- Velvet - сборщик геномов
Биоинформатика и вычислительная биология
Под биоинформатикой понимают любое использование компьютеров для обработки биологической информации. На практике, иногда это определение более узкое, под ним понимают использование компьютеров для обработки экспериментальных данных по структуре биологических макромолекул (белков и нуклеиновых кислот) с целью получения биологически значимой информации. В свете изменения шифра научных специальностей (03.00.28 "Биоинформатика" превратилась в 03.01.09 "Математическая биология, биоинформатика") поле термина "биоинформатика" расширилось и включает все реализации математических алгоритмов, связанных с биологическими объектами.
Термины биоинформатика и «вычислительная биология » часто употребляются как синонимы, хотя последний чаще указывает на разработку алгоритмов и конкретные вычислительные методы. Считается, что не всякое использование вычислительных методов в биологии является биоинформатикой, например, математическое моделирование биологических процессов - это не биоинформатика.
Биоинформатика использует методы прикладной математики , статистики и информатики . Исследования в вычислительной биологии нередко пересекаются с системной биологией . Основные усилия исследователей в этой области направлены на изучение геномов , анализ и предсказание структуры белков , анализ и предсказание взаимодействий молекул белка друг с другом и другими молекулами, а также реконструкция эволюции .
Биоинформатика и её методы используются также в биохимии , биофизике , экологии и в других областях. Основная линия в проектах биоинформатики - это использование математических средств для извлечения полезной информации из «шумных» или слишком объёмных данных о структуре ДНК и белков, полученных экспериментально.
Структурная биоинформатика
К структурной биоинформатике относится разработка алгоритмов и программ для предсказания пространственной структуры белков. Темы исследований в структурной биоинформатике:
- Рентгеноструктурный анализ (РСА) макромолекул
- Индикаторы качества модели макромолекулы, построенной по данным РСА
- Алгоритмы вычисления поверхности макромолекулы
- Алгоритмы нахождения гидрофобного ядра молекулы белка
- Алгоритмы нахождения структурных доменов белков
- Пространственное выравнивание структур белков
- Структурные классификации доменов SCOP и CATH
- Молекулярная динамика
Примечания
См. также
Wikimedia Foundation . 2010 .
Синонимы :Смотреть что такое "Биоинформатика" в других словарях:
Сущ., кол во синонимов: 1 биология (73) Словарь синонимов ASIS. В.Н. Тришин. 2013 … Словарь синонимов
Биоинформатика - (син. Вычислительная биология) биологическая дисциплина, занимающаяся исследованием, разработкой и применением вычислительных методов (в т.ч. компьютерных) и подходов для расширения использования биологических, поведенческих или медицинских… … Официальная терминология
биоинформатика - Раздел биотехнологии, изучает возможности эффективного использования баз данных и сведений, накопленных с помощью функциональной, структурной геномики, комбинаторной химии, скрининга, протеомики и ДНК секвинирования… … Справочник технического переводчика
Биоинформатика - * біяінфарматыка * bioinformatics новое направление исследований, использующее математические и алгоритмические методы для решения молекулярно биологических задач. Задачи Б. можно определить как развитие и использование математических и… …
Биоинформатика - (bioinformatics). Дисциплина, в которой соединились биология, компьютерные технологии и информатика … Психология развития. Словарь по книге
Белковая биоинформатика - * бялковая біяінфарматыка * protein bioinformatics анализ белковых суперсемейств методами биоинформатики и экспериментальными исследованиями для разработки стратегий в области белковой биоинженерии. Этот анализ используется для выяснения роли… … Генетика. Энциклопедический словарь
Бактериальная биоинформатика - * бактэрыяльная біяінфарматыка * bacterial bioinformatics использование компьютерных методов скрининга секвенированных геномов патогенов для разработки антимикробных препаратов. Устойчивость к антибиотикам среди вирулентных видов увеличивается,… … Генетика. Энциклопедический словарь
Клеточная биоинформатика - * клетачная біяінфарматыка * cellular bioinformatics небольшой раздел биоинформатики (см.), сфокусированный на исследовании функционирования живых клеток с привлечением всех имеющихся данных о ДНК, мРНК, белках и процессах метаболизма. Один из… … Генетика. Энциклопедический словарь
Медицинская биоинформатика - * медыцынская біяінфарматыка * medical bioinformatics научная дисциплина, использующая методы биоинформатики (см.) в медицине … Генетика. Энциклопедический словарь
Выделениe ДНК методом спиртового осаждения. ДНК выглядит как клубок белых нитей … Википедия
Если спросить случайного прохожего, что такое биология, он наверняка ответит что-то вроде «наука о живой природе». Про информатику скажет, что она имеет дело с компьютерами и информацией. Если мы не побоимся быть навязчивыми и зададим ему третий вопрос – что такое биоинформатика? – тут-то он наверняка и растеряется. Логично: про эту область знаний даже в ЕРАМ знает далеко не каждый – хотя в нашей компании и биоинформатики есть. Давайте разбираться, для чего эта наука нужна человечеству вообще и ЕРАМ в частности: в конце концов, вдруг нас на улице об этом спросят.
Почему биология перестала справляться без информатики и при чем тут рак
Чтобы провести исследование, биологам уже недостаточно взять анализы и посмотреть в микроскоп. Современная биология имеет дело с колоссальными объемами данных. Часто обработать их вручную просто невозможно, поэтому многие биологические задачи решаются вычислительными методами. Не будем далеко ходить: молекула ДНК настолько мала, что разглядеть ее под световым микроскопом нельзя. А если и можно (под электронным), всё равно визуальное изучение не помогает решить многих задач.ДНК человека состоит из трех миллиардов нуклеотидов – чтобы вручную проанализировать их все и найти нужный участок, не хватит и целой жизни. Ну, может и хватит – одной жизни на анализ одной молекулы – но это слишком долго, дорого и малопродуктивно, так что геном анализируют при помощи компьютеров и вычислений.
Биоинформатика - это и есть весь набор компьютерных методов для анализа биологических данных: прочитанных структур ДНК и белков, микрофотографий, сигналов, баз данных с результатами экспериментов и т. д.

Иногда секвенировать ДНК нужно, чтобы подобрать правильное лечение. Одно и то же заболевание, вызванное разными наследственными нарушениями или воздействием среды, нужно лечить по-разному. А еще в геноме есть участки, которые не связаны с развитием болезни, но, например, отвечают за реакцию на определенные виды терапии и лекарств. Поэтому разные люди с одним и тем же заболеванием могут по-разному реагировать на одинаковое лечение.
Еще биоинформатика нужна, чтобы разрабатывать новые лекарства. Их молекулы должны иметь определенную структуру и связываться с определенным белком или участком ДНК. Смоделировать структуру такой молекулы помогают вычислительные методы.
Достижения биоинформатики широко применяют в медицине, в первую очередь в терапии рака. В ДНК зашифрована информация о предрасположенности и к другим заболеваниям, но над лечением рака работают больше всего. Это направление считается самым перспективным, финансово привлекательным, важным – и самым сложным.
Биоинформатика в ЕРАМ
В ЕРАМ биоинформатикой занимается подразделение Life Sciences. Там разрабатывают программное обеспечение для фармкомпаний, биологических и биотехнологических лабораторий всех масштабов - от стартапов до ведущих мировых компаний. Справиться с такой задачей могут только люди, которые разбираются в биологии, умеют составлять алгоритмы и программировать.Биоинформатики – гибридные специалисты. Сложно сказать, какое знание для них первично: биология или информатика. Если так ставить вопрос, им нужно знать и то и другое. В первую очередь важны, пожалуй, аналитический склад ума и готовность много учиться. В ЕРАМ есть и биологи, которые доучились информатике, и программисты с математиками, которые дополнительно изучали биологию.
Как становятся биоинформатиками
Мария Зуева, разработчик:«Я получила стандартное ИТ-образование, потом училась на курсах ЕРАМ Java Lab, где увлеклась машинным обучением и Data Science. Когда я выпускалась из лаборатории, мне сказали: «Сходи в Life Sciences, там занимаются биоинформатикой и как раз набирают людей». Не лукавлю: тогда я услышала слово «биоинформатика» в первый раз. Прочитала про нее на Википедии и пошла.
Тогда в подразделение набрали целую группу новичков, и мы вместе изучали биоинформатику. Начали с повторения школьной программы про ДНК и РНК, затем подробно разбирали существующие в биоинформатике задачи, подходы к их решению и алгоритмы, учились работать со специализированным софтом».
«По образованию я биофизик, в 2012-м защитил кандидатскую по генетике. Какое-то время работал в науке, занимался исследованиями – и продолжаю до сих пор. Когда появилась возможность применить научные знания в производстве, я тут же за нее ухватился.
Для бизнес-аналитика у меня весьма специфическая работа. Например, финансовые вопросы проходят мимо меня, я скорее эксперт по предметной области. Я должен понять, чего от нас хотят заказчики, разобраться в проблеме и составить высокоуровневую документацию – задание для программистов, иногда сделать работающий прототип программы. По ходу проекта я поддерживаю контакт с разработчиками и заказчиками, чтобы те и другие были уверены: команда делает то, что от нее требуется. Фактически я переводчик с языка заказчиков – биологов и биоинформатиков – на язык разработчиков и обратно».
Как читают геном
Чтобы понять суть биоинформатических проектов ЕРАМ, сначала нужно разобраться, как секвенируют геном. Дело в том, что проекты, о которых мы будем говорить, напрямую связаны с чтением генома. Обратимся за объяснением к биоинформатикам.Михаил Альперович, глава юнита биоинформатики:
«Представьте, что у вас есть десять тысяч экземпляров «Войны и мира». Вы пропустили их через шредер, хорошенько перемешали, наугад вытащили из этой кучи ворох бумажных полосок и пытаетесь собрать из них исходный текст. Вдобавок у вас есть рукопись «Войны и мира». Текст, который вы соберете, нужно будет сравнить с ней, чтобы отловить опечатки (а они обязательно будут). Примерно так же читают ДНК современные машины-секвенаторы. ДНК выделяют из клеточных ядер и делят на фрагменты по 300–500 пар нуклеотидов (мы помним, что в ДНК нуклеотиды связаны друг с другом попарно). Молекулы дробят, потому что ни одна современная машина не может прочитать геном от начала до конца. Последовательность слишком длинная, и по мере ее прочтения накапливаются ошибки.
Вспоминаем «Войну и мир» после шредера. Чтобы восстановить исходный текст романа, нам нужно прочитать и расположить в правильном порядке все кусочки романа. Получается, что мы читаем книгу несколько раз по крошечным фрагментам. То же с ДНК: каждый участок последовательности секвенатор прочитывает с многократным перекрытием – ведь мы анализируем не одну, а множество молекул ДНК.
Полученные фрагменты выравнивают – «прикладывают» каждый из них к эталонному геному и пытаются понять, какому участку эталона соответствует прочитанный фрагмент. Затем в выравненных фрагментах находят вариации – значащие отличия прочтений от эталонного генома (опечатки в книге по сравнению с эталонной рукописью). Этим занимаются программы – вариант-коллеры (от англ. variant caller – выявитель мутаций). Это самая сложная часть анализа, поэтому различных программ – вариант-коллеров много и их постоянно совершенствуют и разрабатывают новые.
Подавляющее большинство найденных мутаций нейтральны и ни на что не влияют. Но есть и такие, в которых зашифрованы предрасположенность к наследственным заболеваниям или способность откликаться на разные виды терапии».

Для анализа берут образец, в котором находится много клеток - а значит, и копий полного набора ДНК клетки. Каждый маленький фрагмент ДНК прочитывают несколько раз, чтобы минимизировать вероятность ошибки. Если пропустить хотя бы одну значащую мутацию, можно поставить пациенту неверный диагноз или назначить неподходящее лечение. Прочитать каждый фрагмент ДНК по одному разу слишком мало: единственное прочтение может быть неправильным, и мы об этом не узнаем. Если мы прочитаем тот же фрагмент дважды и получим один верный и один неверный результат, нам будет сложно понять, какое из прочтений правдивое. А если у нас сто прочтений и в 95 из них мы видим один и тот же результат, мы понимаем, что он и есть верный.
Геннадий Захаров:
«Для анализа раковых заболеваний секвенировать нужно и здоровую, и больную клетку. Рак появляется в результате мутаций, которые клетка накапливает в течение своей жизни. Если в клетке испортились механизмы, отвечающие за ее рост и деление, то клетка начинает неограниченно делиться вне зависимости от потребностей организма, т. е. становится раковой опухолью. Чтобы понять, чем именно вызван рак, у пациента берут образец здоровой ткани и раковой опухоли. Оба образца секвенируют, сопоставляют результаты и находят, чем один отличается от другого: какой молекулярный механизм сломался в раковой клетке. Исходя из этого подбирают лекарство, которое эффективно против клеток с “поломкой”».
Биоинформатика: производство и опенсорс
У подразделения биоинформатики в ЕРАМ есть и производственные, и опенсорс-проекты. Причем часть производственного проекта может перерасти в опенсорс, а опенсорсный проект – стать частью производства (например, когда продукт ЕРАМ с открытым кодом нужно интегрировать в инфраструктуру клиента).Проект №1: вариант-коллер
Для одного из клиентов – крупной фармацевтической компании – ЕРАМ модернизировал программу вариант-коллер. Ее особенность в том, что она способна находить мутации, недоступные другим аналогичным программам. Изначально программа была написана на языке Perl и обладала сложной логикой. В ЕРАМ программу переписали на Java и оптимизировали – теперь она работает в 20, если не в 30 раз быстрее.Исходный код программы доступен на GitHub .
Проект №2: 3D-просмотрщик молекул
Для визуализации структуры молекул в 3D есть много десктоп- и веб-приложений. Представлять, как молекула выглядит в пространстве, крайне важно, например, для разработки лекарств. Предположим, нам нужно синтезировать лекарство, обладающее направленным действием. Сначала нам потребуется спроектировать молекулу этого лекарства и убедиться, что она будет взаимодействовать с нужными белками именно так, как нужно. В жизни молекулы трехмерные, поэтому анализируют их тоже в виде трехмерных структур.Для 3D-просмотра молекул ЕРАМ сделал онлайн-инструмент, который изначально работал только в окне браузера. Потом на основании этого инструмента разработали версию, которая позволяет визуализировать молекулы в очках виртуальной реальности HTC Vive. К очкам прилагаются контроллеры, которыми молекулу можно поворачивать, перемещать, подставлять к другой молекуле, поворачивать отдельные части молекулы. Делать всё это в 3D куда удобнее, чем на плоском мониторе. Эту часть проекта биоинформатики ЕРАМ делали совместно с подразделением Virtual Reality, Augmented Reality and Game Experience Delivery.
Программа только готовится к публикации на GitHub, зато пока есть , по которой можно посмотреть ее демо-версию.
Как выглядит работа с приложением, можно узнать из видео .
Проект №3: геномный браузер NGB
Геномный браузер визуализирует отдельные прочтения ДНК, вариации и другую информацию, сгенерированную утилитами для анализа генома. Когда прочтения сопоставлены с эталонным геномом и мутации найдены, ученому остается проконтролировать, правильно ли сработали машины и алгоритмы. От того, насколько точно выявлены мутации в геноме, зависит, какой диагноз поставят пациенту или какое лечение ему назначат. Поэтому в клинической диагностике контролировать работу машин должен ученый, а помогает ему в этом геномный браузер.Биоинформатикам-разработчикам геномный браузер помогает анализировать сложные случаи, чтобы найти ошибки в работе алгоритмов и понять, как их можно улучшить.
Новый геномный браузер NGB (New Genome Browser) от ЕРАМ работает в вебе, но по скорости и функционалу не уступает десктопным аналогам. Это продукт, которого не хватало на рынке: предыдущие онлайновые инструменты работали медленнее и умели делать меньше, чем десктопные. Сейчас многие клиенты выбирают веб-приложения из соображений безопасности. Онлайн-инструмент позволяет ничего не устанавливать на рабочий компьютер ученого. С ним можно работать из любой точки мира, зайдя на корпоративный портал. Ученому не обязательно всюду возить за собой рабочий компьютер и скачивать на него все необходимые данные, которых может быть очень много.

Геннадий Захаров, бизнес-аналитик:
«Над опенсорсными утилитами я работал частично как заказчик: ставил задачу. Я изучал лучшие решения на рынке, анализировал их преимущества и недостатки, искал, как можно их усовершенствовать. Нам нужно было сделать веб-решения не хуже десктопных аналогов и при этом добавить в них что-то уникальное.
В 3D-просмотрщике молекул это была работа с виртуальной реальностью, а в геномном браузере – улучшенная работа с вариациями. Мутации бывают сложными. Перестройки в раковых клетках иногда затрагивают огромные области. В них появляются лишние хромосомы, куски хромосом и целые хромосомы исчезают или объединяются в случайном порядке. Отдельные куски генома могут копироваться по 10–20 раз. Такие данные, во-первых, сложнее получить из прочтений, а во-вторых, сложнее визуализировать.
Мы разработали визуализатор, который правильно читает информацию о таких протяженных структурных перестройках. Еще мы сделали набор визуализаций, который при контакте хромосом показывает, образовались ли из-за этого контакта гибридные белки. Если протяженная вариация затрагивает несколько белков, мы по клику можем рассчитать и показать, что происходит в результате такой вариации, какие гибридные белки получаются. В других визуализаторах ученым приходилось отслеживать эту информацию вручную, а в NGB – в один клик».
Как изучать биоинформатику
Мы уже говорили, что биоинформатики – гибридные специалисты, которые должны знать и биологию, и информатику. Самообразование играет в этом не последнюю роль. Конечно, в ЕРАМ есть вводный курс в биоинформатику, но рассчитан он на сотрудников, которым эти знания пригодятся на проекте. Занятия проводятся только в Санкт-Петербурге. И всё же, если биоинформатика вам интересна, возможность учиться есть:Профессия - биоинформатик
Что это такое?
Информатика – отрасль науки, изучающая структуру и общие свойства информации, а также вопросы, связанные с ее сбором, хранением, поиском, переработкой, преобразованием, распространением и использованием в различных сферах деятельности. Биоинформатикой же называют информатику в применении к молекулярной биологии.
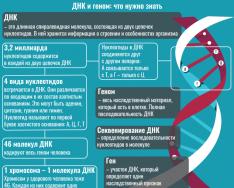
Все знают, что прочитан геном человека. Что такое геном с точки зрения информатики? Это длинный текст, содержащий около 3 млрд букв (нуклеотидов A, T, G, C). И все. Одной из проблем биоинформатики является установление смысла этого текста.
Разумеется, кроме самой последовательности ДНК есть много дополнительной экспериментальной информации.
Далеко не все гены человека известны, о функциях многих генов нет данных. Задача биоинформатики заключается в том, чтобы найти ранее неизвестные гены и описать их предположительную функцию. Как ищутся гены? Это трудная задача. Здесь на помощь приходит математика. В гигантском массиве информации с помощью современных математических методов ищутся скрытые закономерности, которые и позволяют находить гены и предсказывать их свойства.
Говоря о геноме, обычно проводят аналогию с расшифровкой древних рукописей, когда текст известен, а язык - нет. Эта задача неразрешима до тех пор, пока у нас нет никаких представлений о содержании текста. Однако, если мы хотя бы примерно представляем, о чем этот текст, то появляется надежда на его осмысление. В биоинформатике ситуация лучше, чем при расшифровке древних письмен, поскольку ее предсказания могут быть проверены экспериментально.
Гены кодируют белки, поэтому предсказание функции гена - это то же самое, что предсказание функции белка. Для многих белков функции известны из эксперимента. Используя эти данные, метод аналогий и другие методы современной математики, иногда удается предсказать функции других белков.
Сейчас в современных лабораториях часто используют технику массовых экспериментов, когда в одном опыте получают информацию о тысячах генов. Разобраться в этом море информации можно только с помощью компьютера. Проект «Геном человека» - типичный пример такого подхода. Другой пример. Если определить активность всех генов в здоровой и раковой клетке, то после анализа данных можно узнать, какие гены отвечают за перерождение здоровой клетки в раковую. Все было бы просто, если бы такие экспериментальные данные не содержали в себе очень много шума, т.е. ошибок.
Гены - это последовательности ДНК, белки - это аминокислотные последовательности. Функциональность белков определяется их пространственной формой. При этом белки, имеющие разные аминокислотные последовательности, могут иметь очень похожую пространственную структуру. Одной из классических (и до сих пор не решенных) задач биоинформатики является предсказание пространственной структуры белка по последовательности аминокислот. Уже более 5 лет существуют международные соревнования методов предсказания пространственной структуры белка по его последовательности.
Почему это интересно?
Анализ геномов приносит множество новой информации. В настоящее время расшифровано более 200 геномов различных бактерий, каждый из которых содержит несколько тысяч генов. Для того чтобы охарактеризовать один ген, требуется несколько месяцев напряженной работы экспериментаторов. С другой стороны, для того чтобы достаточно подробно описать один бактериальный геном средствами биоинформатики, достаточно примерно месяца работы небольшой группы исследователей.
В геноме человека около 35 тыс. генов (всего в 10 раз больше, чем у бактерии, и в 2 раза больше, чем у плодовой мушки), а количество синтезируемых белков гораздо больше. В чем же дело? Оказывается, что очень часто один ген кодирует несколько разных форм белка. За это отвечает явление, названное альтернативным сплайсингом . Биоинформатика впервые показала, что количество генов, имеющих альтернативный сплайсинг, очень велико. Осталось загадкой, как все это регулируется.
В клетке не все гены должны работать одновременно. Для того чтобы гены работали, как слаженный оркестр, необходимо, чтобы гены включались только тогда, когда их работа необходима. Этим заведует система регуляции генов, анализ которой позволил обнаружить принципиально новые способы регуляции – рибопереключатели .
Еще одно направление - исследование эволюции всего живого. Здесь тоже есть много открытий, например горизонтальный перенос генов между видами. Биоинформатика в некоторых случаях позволяет не только показать эти случаи, но также и датировать их.
Зачем это нужно?
Биология и биоинформатика являются не только способами познания мира, но имеют и прикладное значение, прежде всего в медицине и биотехнологии.
Биоинформатика играет существенную роль в поиске новых лекарственных препаратов и мишеней для них, а также в отбраковке неперспективных лекарств. Приведу пример.
Все вы слышали про мыло Safeguard, которое убивает микробы. Оказалось, что есть весьма опасные стрептококки, не чувствительные к его действующему началу - триклозану. Сначала это было показано с помощью компьютерного анализа геномов стрептококков, а потом подтверждено экспериментально.
 Еще пример - анализ генетических данных
людей здоровых и с каким-либо заболеванием,
например ишемической болезнью сердца. Нет одного
гена, ответственного за эту болезнь. Однако
сопоставление данных по большому количеству
больных позволило найти так называемые
ассоциации - набор генов предрасположенности к
указанной болезни, и тем самым дает возможность
определить генетическую группу риска.
Еще пример - анализ генетических данных
людей здоровых и с каким-либо заболеванием,
например ишемической болезнью сердца. Нет одного
гена, ответственного за эту болезнь. Однако
сопоставление данных по большому количеству
больных позволило найти так называемые
ассоциации - набор генов предрасположенности к
указанной болезни, и тем самым дает возможность
определить генетическую группу риска.
Биоинформатика широко используется в биотехнологии, задачу которой в общем виде можно сформулировать как получение как можно большего количества целевого продукта из 1 г, например, сахара. Для этого надо детально изучить пути биосинтеза, исследовать систему регуляции, найти в других организмах более эффективные ферменты. Здесь тоже всю подготовительную работу может взять на себя биоинформатика.
Важность этого направления науки можно показать и косвенно. Достаточно сказать, что в мире есть несколько крупных научных биоинформатических центров, есть коммерческие компании, предоставляющие биоинформатические услуги. Любая крупная или средняя фармацевтическая или биотехнологическая компания имеет отдел биоинформатики. Сейчас многие университеты готовят специалистов в этой области. В нашей стране возрождается фармацевтическая и биотехнологическая промышленность, которой в скором времени потребуются специалисты. Академическая наука также нуждается в грамотных биоинформатиках.
Что надо знать и уметь?
Грамотный биоинформатик должен иметь разностороннее образование. Он должен хорошо знать биологию. Кроме того, он должен владеть многими методами математики: статистикой, теорией вероятностей, вычислительной математикой, теорией алгоритмов. Надо знать физику и химию - чтобы не делать глупостей. Необходимо знать английский язык - чтобы читать научную литературу. Надо постоянно интересоваться новыми результатами как в биоинформатике, так и в биологии в целом.
В общем, надо быть культурным человеком и постоянно стремиться узнавать что-нибудь новое.
Физика